Advanced calcium-blocking technology*1


SAPIEN 3 Ultra valve, powered by RESILIA tissue technology
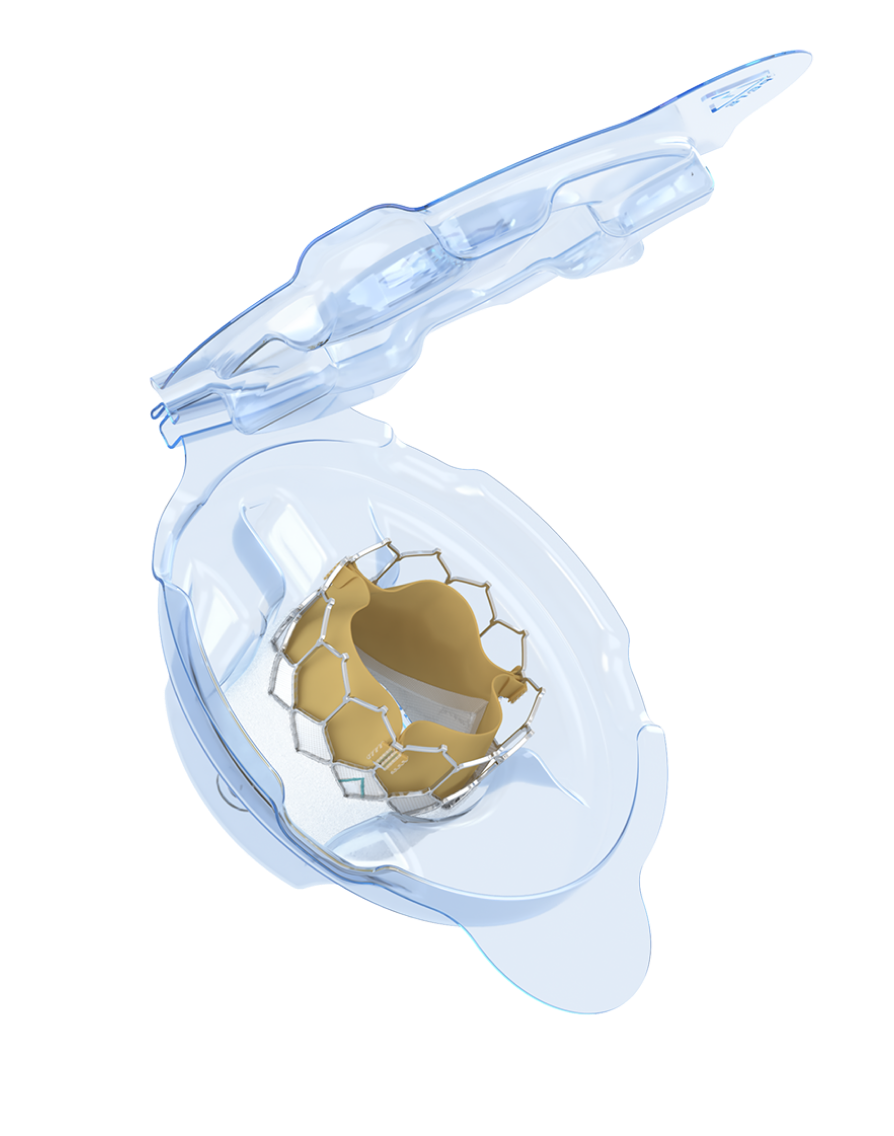
Advanced calcium-blocking technology*1

Three independently matched leaflets for thickness and elasticity

Potential to improve valve longevity and reduce reintervention*†1

The only transcatheter heart valve (THV) with dry tissue storage
*Clinical data on surgical valves with RESILIA tissue up to 7-year follow-up have been published, with additional follow-up to 10-years in progress.
†RESILIA tissue tested against tissue from commercially available bovine pericardial from Edwards Lifesciences in a juvenile sheep model. Flameng, et al. J Thorac Cardiovasc Surg.
Read the latest clinical trial results for aortic valve replacement with RESILIA tissue
Indications: The Edwards SAPIEN 3, SAPIEN 3 Ultra, and SAPIEN 3 Ultra RESILIA Transcatheter Heart Valve system is indicated to reduce the risks associated with progression from asymptomatic to symptomatic severe native calcific aortic stenosis in patients who are judged by a heart team to be appropriate for transcatheter heart valve replacement therapy.
The Edwards SAPIEN 3, SAPIEN 3 Ultra, and SAPIEN 3 Ultra RESILIA Transcatheter Heart Valve system is indicated for relief of aortic stenosis in patients with symptomatic heart disease due to severe native calcific aortic stenosis who are judged by a Heart Team, including a cardiac surgeon, to be appropriate for the transcatheter heart valve replacement therapy.
The Edwards SAPIEN 3, SAPIEN 3 Ultra, and SAPIEN 3 Ultra RESILIA Transcatheter Heart Valve system is indicated for patients with symptomatic heart disease due to a failing (stenosed, insufficient, or combined) surgical or transcatheter bioprosthetic aortic valve, or a native mitral valve with an annuloplasty ring who are judged by a heart team, including a cardiac surgeon, to be at high or greater risk for open surgical therapy (i.e., predicted risk of surgical mortality ≥ 8% at 30 days, based on the Society of Thoracic Surgeons (STS) risk score and other clinical co-morbidities unmeasured by the STS risk calculator).
The Edwards SAPIEN 3, SAPIEN 3 Ultra, and SAPIEN 3 Ultra RESILIA Transcatheter Heart Valve system is indicated for patients with symptomatic heart disease due to a failing (stenosed, insufficient, or combined) surgical bioprosthetic mitral valve who are judged by a heart team, including a cardiac surgeon, to be at intermediate or greater risk for open surgical therapy (i.e., predicted risk of surgical mortality ≥ 4% at 30 days, based on the Society of Thoracic Surgeons (STS) risk score and other clinical co-morbidities unmeasured by the STS risk calculator).
Contraindications: The valves and delivery systems are contraindicated in patients who cannot tolerate an anticoagulation/antiplatelet regimen or who have active bacterial endocarditis or other active infections, or who have significant annuloplasty ring dehiscence.
Warnings: Observation of the pacing lead throughout the procedure is essential to avoid the potential risk of pacing lead perforation. There may be an increased risk of stroke in transcatheter aortic valve replacement procedures, as compared to balloon aortic valvuloplasty or other standard treatments in high or greater risk patients. The devices are designed, intended, and distributed for single use only. Do not resterilize or reuse the devices. There are no data to support the sterility, nonpyrogenicity, and functionality of the devices after reprocessing. Incorrect sizing of the valve may lead to paravalvular leak, migration, embolization, residual gradient (patient-prosthesis mismatch), and/or annular rupture. Accelerated deterioration of the valve due to calcific degeneration may occur in children, adolescents, or young adults and in patients with an altered calcium metabolism. Prior to delivery, the valve must remain hydrated at all times and cannot be exposed to solutions other than its shipping storage solution and sterile physiologic rinsing solution. Valve leaflets mishandled or damaged during any part of the procedure will require replacement of the valve. Caution should be exercised in implanting a valve in patients with clinically significant coronary artery disease. Patients with pre-existing prostheses should be carefully assessed prior to implantation of the valve to ensure proper valve positioning and deployment. Do not use the valve if the tamper-evident seal is broken or the storage solution does not completely cover the valve (SAPIEN 3 and SAPIEN 3 Ultra only), the temperature indicator has been activated, the valve is damaged, or the expiration date has elapsed. Do not mishandle the delivery system or use it if the packaging or any components are not sterile, have been opened or are damaged (e.g., kinked or stretched), or if the expiration date has elapsed. Use of excessive contrast media may lead to renal failure. Measure the patient’s creatinine level prior to the procedure. Contrast media usage should be monitored. Patient injury could occur if the delivery system is not un-flexed prior to removal. Care should be exercised in patients with hypersensitivities to cobalt, nickel, chromium, molybdenum, titanium, manganese, silicon, and/or polymeric materials. The procedure should be conducted under fluoroscopic guidance. Some fluoroscopically guided procedures are associated with a risk of radiation injury to the skin. These injuries may be painful, disfiguring, and long-lasting. Valve recipients should be maintained on anticoagulant/antiplatelet therapy, except when contraindicated, as determined by their physician. This device has not been tested for use without anticoagulation. Do not add or apply antibiotics to the storage solution (SAPIEN 3 and SAPIEN 3 Ultra only), rinse solution, or to the valve. Balloon valvuloplasty should be avoided in the treatment of failing bioprostheses as this may result in embolization of bioprosthesis material and mechanical disruption of the valve leaflets. Do not perform stand-alone balloon aortic valvuloplasty procedures in the INSPIRIS RESILIA aortic valve for the sizes 19-25 mm. This may expand the valve causing aortic incompetence, coronary embolism or annular rupture. Transcatheter valve replacement in mitral annuloplasty rings is not recommended in cases of partial annuloplasty ring dehiscence due to high risk of PVL. Transcatheter valve replacement in mitral annuloplasty rings is not recommended in cases of partial (incomplete) annuloplasty rings in the absence of annular calcium due to increased risk of valve embolization. Transcatheter valve replacement in mitral annuloplasty rings is not recommended in cases of rigid annuloplasty rings due to increased risk of PVL or THV deformation.
Precautions: Long-term durability has not been established for the valve. Regular medical follow-up is advised to evaluate valve performance. Limited clinical data are available for transcatheter aortic valve replacement in patients with a congenital bicuspid aortic valve who are deemed to be at low surgical risk. Anatomical characteristics should be considered when using the valve in this population. In addition, patient age should be considered as long-term durability of the valve has not been established. Data on TAVR in patients with asymptomatic severe aortic stenosis are based on study of predominantly low surgical risk patients. Limited clinical data to inform benefit-risk considerations are available for TAVR in patients with asymptomatic severe aortic stenosis who are deemed to be at intermediate or greater surgical risk. Glutaraldehyde may cause irritation of the skin, eyes, nose, and throat. Avoid prolonged or repeated exposure to, or breathing of, the solution. Use only with adequate ventilation. If skin contact occurs, immediately flush the affected area with water; in the event of contact with eyes, seek immediate medical attention. For more information about glutaraldehyde exposure, refer to the Safety Data Sheet available from Edwards Lifesciences. If a significant increase in resistance occurs when advancing the catheter through the vasculature, stop advancement and investigate the cause of resistance before proceeding. Do not force passage, as this could increase the risk of vascular complications. As compared to SAPIEN 3, system advancement force may be higher with the use of SAPIEN 3 Ultra/SAPIEN 3 Ultra RESILIA THV in tortuous/challenging vessel anatomies. To maintain proper valve leaflet coaptation, do not overinflate the deployment balloon. Appropriate antibiotic prophylaxis is recommended post-procedure in patients at risk for prosthetic valve infection and endocarditis. Additional precautions for transseptal replacement of a failed mitral valve bioprosthesis include, the presence of devices or thrombus or other abnormalities in the caval vein precluding safe transvenous femoral access for transseptal approach; and the presence of an Atrial Septal Occluder Device or calcium preventing safe transseptal access. Special care must be exercised in mitral valve replacement to avoid entrapment of the subvalvular apparatus. Safety and effectiveness have not been established for patients with the following characteristics/comorbidities: non-calcified aortic annulus; severe ventricular dysfunction with ejection fraction < 20%; congenital unicuspid aortic valve; pre-existing prosthetic ring in the tricuspid position; severe mitral annular calcification (MAC); severe (> 3+) mitral insufficiency, or Gorlin syndrome; blood dyscrasias defined as leukopenia (WBC < 3000 cells/mL), acute anemia (Hb < 9 g/dL), thrombocytopenia (platelet count < 50,000 cells/mL), or history of bleeding diathesis or coagulopathy; hypertrophic cardiomyopathy with or without obstruction (HOCM); echocardiographic evidence of intracardiac mass, thrombus, or vegetation; a known hypersensitivity or contraindication to aspirin, heparin, ticlopidine (Ticlid), or clopidogrel (Plavix), or sensitivity to contrast media, which cannot be adequately premedicated; significant aortic disease, including abdominal aortic or thoracic aneurysm defined as maximal luminal diameter 5 cm or greater, marked tortuosity (hyperacute bend), aortic arch atheroma (especially if thick [> 5 mm], protruding, or ulcerated) or narrowing (especially with calcification and surface irregularities) of the abdominal or thoracic aorta, severe “unfolding” and tortuosity of the thoracic aorta; access characteristics that would preclude safe placement of the Edwards sheath, such as severe obstructive calcification or severe tortuosity; bulky calcified aortic valve leaflets in close proximity to coronary ostia; a concomitant paravalvular leak where the failing prosthesis is not securely fixed in the native annulus or is not structurally intact (e.g., wireform frame fracture, annuloplasty ring dehiscence); or a partially detached leaflet of the failing bioprosthesis that in the aortic position may obstruct a coronary ostium. For Left axillary approach, a left subclavian takeoff angle ~ ≥ 90° from the aortic arch causes sharp angles, which may be responsible for potential sheath kinking, subclavian/axillary dissection and aortic arch damage. For left/right axillary approach, ensure there is flow in Left Internal Mammary Artery (LIMA)/Right Internal Mammary Artery (RIMA) during procedure and monitor pressure in homolateral radial artery. Residual mean gradient may be higher in a “THV-in-failing prosthesis” configuration than that observed following implantation of the valve inside a native aortic annulus using the same size device. Patients with elevated mean gradient post procedure should be carefully followed. It is important that the manufacturer, model and size of the preexisting prosthesis be determined, so that the appropriate valve can be implanted and a prosthesis-patient mismatch be avoided. Additionally, pre-procedure imaging modalities must be employed to make as accurate a determination of the inner diameter as possible.
Potential Adverse Events: Potential risks associated with the overall procedure, including potential access complications associated with standard cardiac catheterization, balloon valvuloplasty, the potential risks of conscious sedation and/or general anesthesia, and the use of angiography: death; stroke/transient ischemic attack, clusters, or neurological deficit; paralysis; permanent disability; respiratory insufficiency or respiratory failure; hemorrhage requiring transfusion or intervention; cardiovascular injury including perforation or dissection of vessels, ventricle, atrium, septum, myocardium, or valvular structures that may require intervention; pericardial effusion or cardiac tamponade; thoracic bleeding; embolization including air, calcific valve material, or thrombus; infection including septicemia and endocarditis; heart failure; myocardial infarction; renal insufficiency or renal failure; conduction system defect which may require a permanent pacemaker; arrhythmia; retroperitoneal bleed; arteriovenous (AV) fistula or pseudoaneurysm; reoperation; ischemia or nerve injury or brachial plexus injury; restenosis; pulmonary edema; pleural effusion; bleeding; anemia; abnormal lab values (including electrolyte imbalance); hypertension or hypotension; allergic reaction to anesthesia, contrast media, or device materials; hematoma; syncope; pain or changes (e.g., wound infection, hematoma, and other wound care complications) at the access site; exercise intolerance or weakness; inflammation; angina; heart murmur; and fever. Additional potential risks associated with the use of the valve, delivery system, and/or accessories include: cardiac arrest; cardiogenic shock; emergency cardiac surgery; cardiac failure or low cardiac output; coronary flow obstruction/transvalvular flow disturbance; device thrombosis requiring intervention; valve thrombosis; device embolization; device migration or malposition requiring intervention; left ventricular outflow tract obstruction; valve deployment in unintended location; valve stenosis; structural valve deterioration (wear, fracture, calcification, leaflet tear/tearing from the stent posts, leaflet retraction, suture line disruption of components of a prosthetic valve, thickening, stenosis); device degeneration; paravalvular or transvalvular leak; valve regurgitation; hemolysis; device explants; nonstructural dysfunction; mechanical failure of delivery system and/or accessories; and non-emergent reoperation.
Indications: The Edwards crimper is indicated for use in preparing the Edwards SAPIEN 3 transcatheter heart valve, Edwards SAPIEN 3 Ultra transcatheter heart valve, and the Edwards SAPIEN 3 Ultra RESILIA transcatheter heart valve for implantation.
Contraindications: There are no known contraindications.
Warnings: The device is designed, intended, and distributed for single use only. Do not resterilize or reuse the device. There are no data to support the sterility, nonpyrogenicity, and functionality of the device after reprocessing. Do not mishandle the device. Do not use the device if the packaging or any components are not sterile, have been opened or are damaged, or the expiration date has elapsed.
Precautions: For special considerations associated with the use of the Edwards crimper prior to THV implantation, refer to the THV Instructions for Use.
Potential Adverse Events: There are no known potential adverse events associated with the Edwards crimper.
CAUTION: Federal (United States) law restricts these devices to sale by or on the order of a physician.